



About M1
M1, the AI-driven intelligent computing platform, is independently developed by Galixir for drug discovery. Combining AI with classical physics principles, M1 makes breakthroughs in classical molecular modeling methods and accurately describes the interaction between molecules and proteins. It calculates the binding free energy between target drug molecules and specific targets with high efficiency and accuracy, and reaches the highest level in the world.
Molecular Docking Module
The high-throughput docking module developed from TBind, Galixir's original AI prediction model of conformational affinity between small molecules and target proteins, designed for M1 platform
GDock predicts the three-dimensional complex structure from the two-dimensional small molecule structure with high efficiency and accuracy beyond classical docking software. This is a huge breakthrough in the field of structure-based drug design. M1's high-throughput docking module, GDock, is capable to rank hudred billions of small molecules based on predicted binding affinity. Meanwhile, GDock also provides reliable input structures for downstream FEP task, further improving the accuracy of binding affinity prediction.
Force Field Module
The next-generation general force field independently developed by Galixir for small molecules, which integrates artificial intelligence technology and classical computational chemistry
With multitudinous quantum mechanics calculation and experimental physicochemical properties as fitting targets, GFF can accurately describe intermolecular and intramolecular interactions, which lays the important groundwork for high-precision molecular simulation and plays a key role in structure-based drug design methods.
Free Energy Perturbation Module
The free energy perturbation computing module on the M1 platform based on artificial intelligence and classical molecular dynamics, which integrates Galixir’s self-developed force field GFF and docking module GDock
GFEP overcomes two major problems in the current FEP calculation: the insufficient validity of input structure and the poor accuracy of the force field. It can calculate the binding free energy between drug molecules and specific targets with high efficiency and accuracy and evaluate the efficacy of drug molecules, thus raising efficiency in the very sinews of drug discovery.

The high-throughput docking module developed from TBind, Galixir's original AI prediction model of conformational affinity between small molecules and target proteins, designed for M1 platform
GDock is the first deep representation learning molecular docking method that can simultaneously predict the three-dimensional binding conformation and binding affinity of small molecules and target proteins. Its performance exceeds the best result of the state-of-the-art method.
GDock is trained in an end-to-end paradigm using graph neural networks of physical heuristics, along with the dual prediction of the three-dimensional complex binding mode and binding affinity. Thence, GDock presents the first breakthrough scheme for molecule-protein interaction prediction in China for its performance surpasses the accuracy and efficiency of the commercial molecular docking software at the international level.
GDock increases the proportion of Ligand RMSD which is less than 5Å from about 30% to 65%; and the proportion of centroid distance based on prediction and reality which is less than 5Å from 48% to 82%.
GDock’s performance on prediction and screening has been improved a lot since it abandons cumbersome classical sampling methods and uses data-driven AI potential energy for structure generation. The global docking task takes only 0.5 seconds for each molecule, which is 1/400th of that of general academic software and 1/2000th of that of well-known commercial docking software. Showing decided superiority in speed and accuracy, GDock is also good at capturing allosteric sites and can tackle allosteric problems that classical Docking software cannot solve.

High-throughput docking

Allosteric Site Identification
GDock has a stronger global prediction ability and excels in capturing the information of allosteric pockets:
PRMT5 protein has multiple binding pockets, and its newly released PDB eutectic structure 6K1S has been found a new binding site. Though GDock has never seen a small molecule binding to this kind of allosteric pocket, it still correctly locates the real binding location (while other methods prefer common orthosteric sites).
Centroid Distance (Å) - global docking
GDock: 0.65
Academic Docking Software: 11.21
Commercial Docking Software: 27.36

GDynamic is an enhanced, production-grade version of the original DynamicBind model, with improved accuracy and expanded functionality. GDynamic is capable of modeling dynamic conformation changes in proteins during docking, which allows it to identify the most suitable protein conformation for the input small-molecule, making the discovery of cryptic pockets possible.

The next-generation general force field independently developed by Galixir for small molecules, which integrates artificial intelligence technology and classical computational chemistry

The classical force field defines atoms in different chemical environments at the abstract level based on expert experience. In order to further enhance GFF to describe the larger chemical space, Galixir uses a graph neural network to construct continuous atom types with AI technology. The whole process is constructed by smooth neural network functions, and the model parameters are end-to-end differentiable. GFF can be easily extended to any molecule on the force field constructed by a graph neural network, making a breakthrough at the classical force field in core modules including energy prediction and conformation optimization. GFF embraces high accuracy, strong generalization and excellent iterative adaptation ability.
AI technology advances the fitting of non-bonded parameters, and GCharge, a charge model based on graph neural network training, is a case in point. Compared with classical non-QM methods, it can obtain charge data close to QM accuracy more quickly and accurately, and improve the prediction accuracy of hydration free energy and small molecule binding free energy of the force field on account of millions of QM calculation data as a training set.
- Energy Prediction
- Conformation Optimization
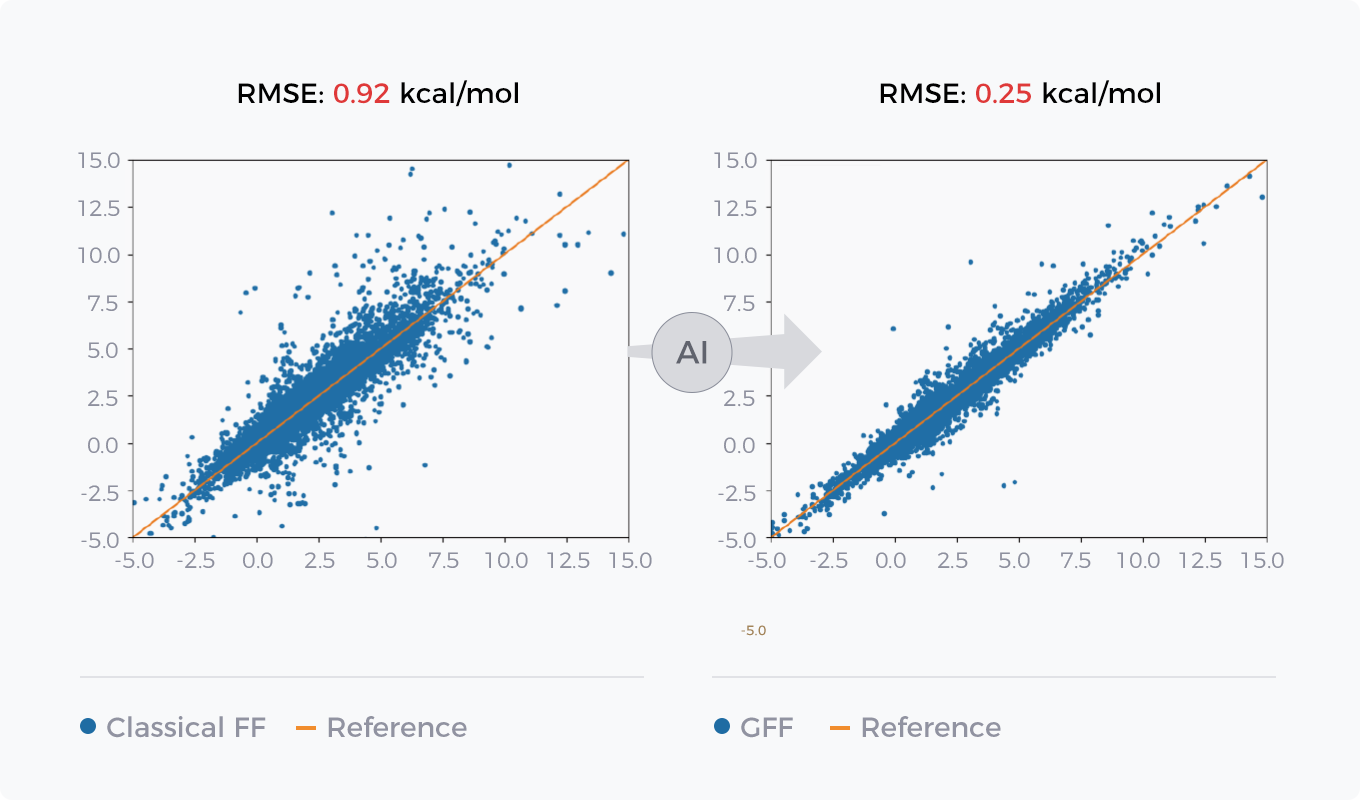
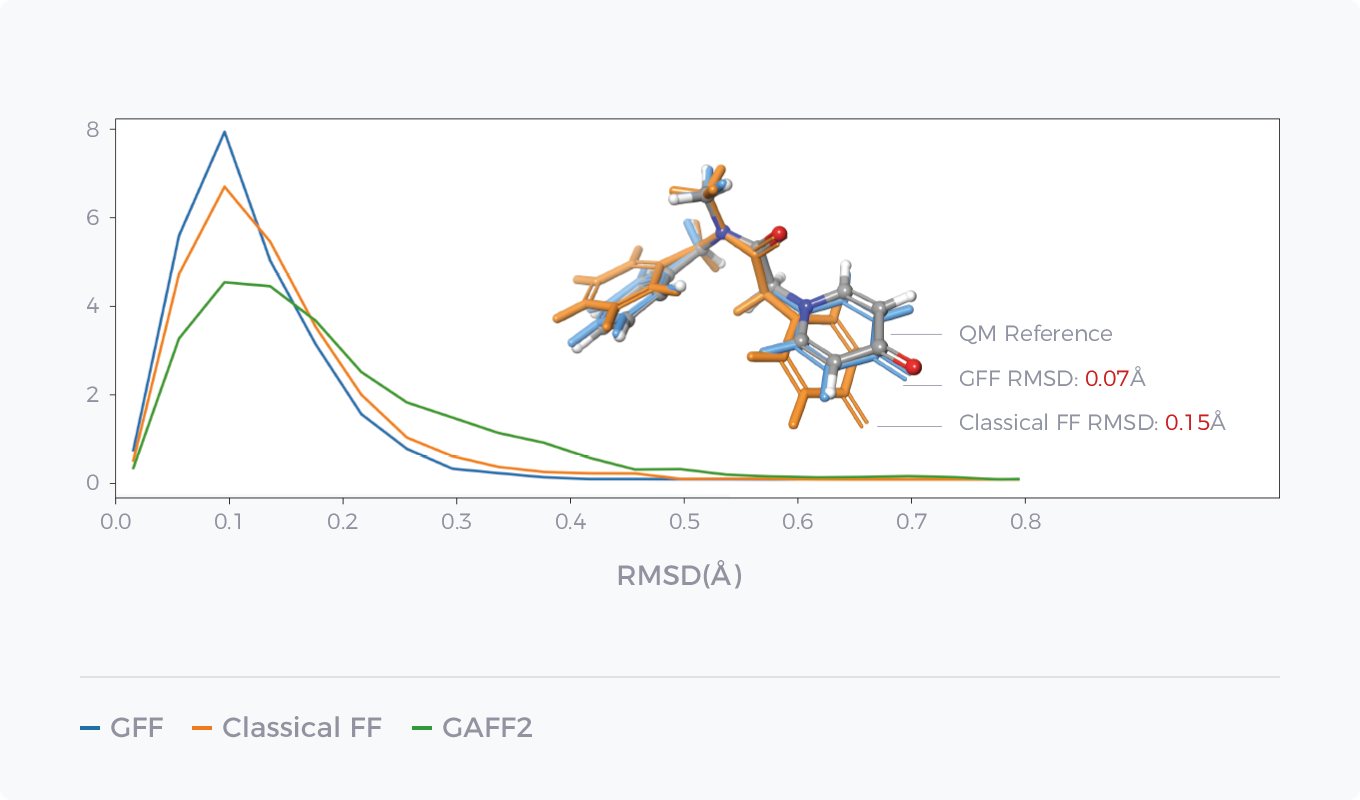
Comparison between GFF of AI force field GFF and Classical Force Field in Energy Prediction and Conformation Optimization
AI technology advances the fitting of non-bonded parameters, and GCharge, a charge model based on graph neural network training, is a case in point. Compared with classical non-QM methods, it can obtain charge data close to QM accuracy more quickly and accurately, and improve the prediction accuracy of hydration free energy and small molecule binding free energy of the force field on account of millions of QM calculation data as a training set.
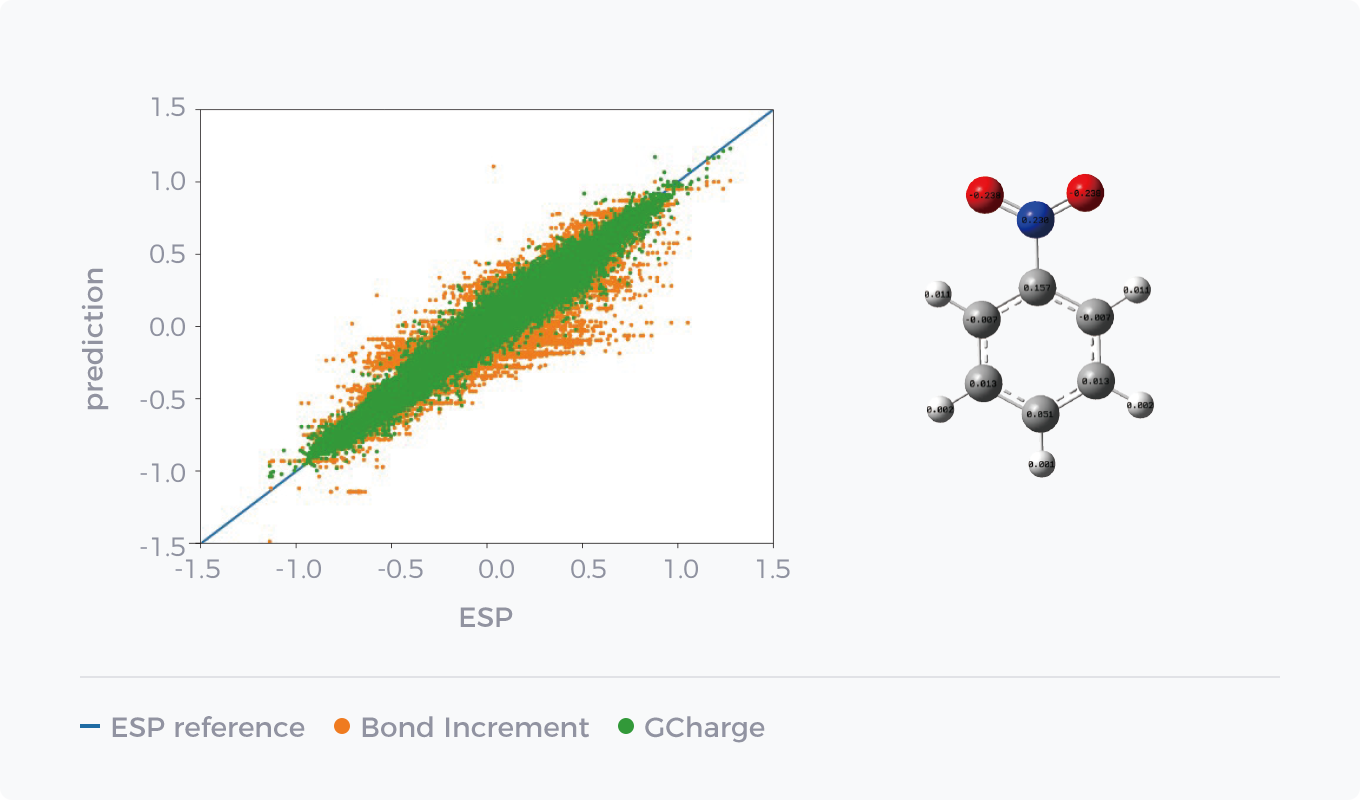
Comparison between GCharge and Classical Charge Model
- Hydration Free Energy Prediction
- Binding Free Energy Prediction
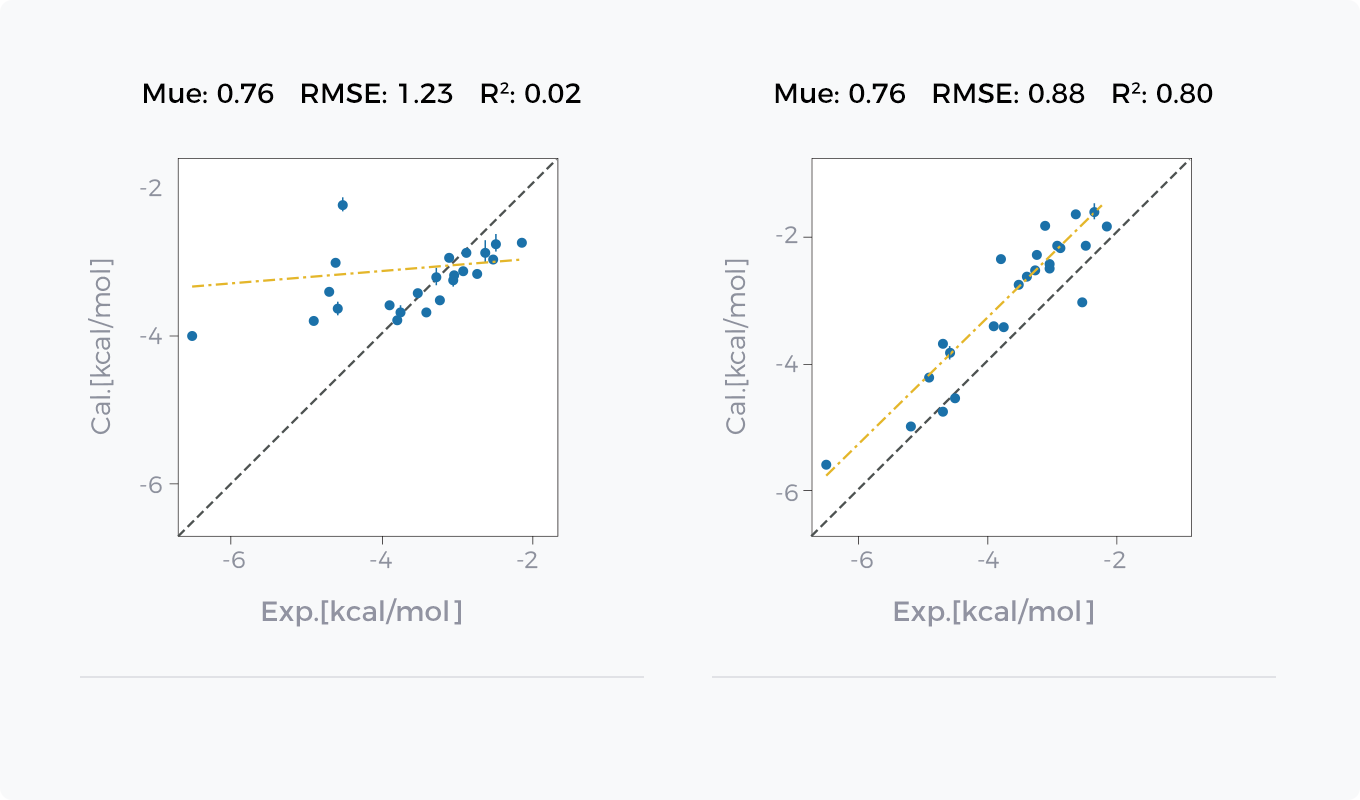

Comparison between GCharge (right) and Classical Charge Model (left) in Hydration Free Energy Prediction and Binding Free Energy Prediction
GFF sets the pace for the industry for it shows high accuracy in many aspects including energy prediction, structural optimization, potential energy surface description, physicochemical property prediction, and free energy calculation, and is superior to most public and commercial force fields.

The free energy perturbation computing module on the M1 platform based on artificial intelligence and classical molecular dynamics, which integrates Galixir’s self-developed force field GFF and docking module GDock
GFEP, an AI-driven free energy perturbation module, is a one-stop computing platform originated by Galixir for FEP calculation requirements in drug discovery. GFEP can automatically complete pretreatment steps of molecular dynamics (MD) simulation like structure optimization, FEP network graph construction, atom mapping, molecular docking, simulation establishment and force field parameter assignment, provided users’ input of protein and ligand information. All simulation calculations will be submitted to the supercomputing cloud platform through the channel built inside Galixir, and scalable computing resources will be scheduled to perform computing tasks for efficiency and data security. Finally, a detailed analysis will be reported on aspects of free energy prediction, interaction, and torsion angles.
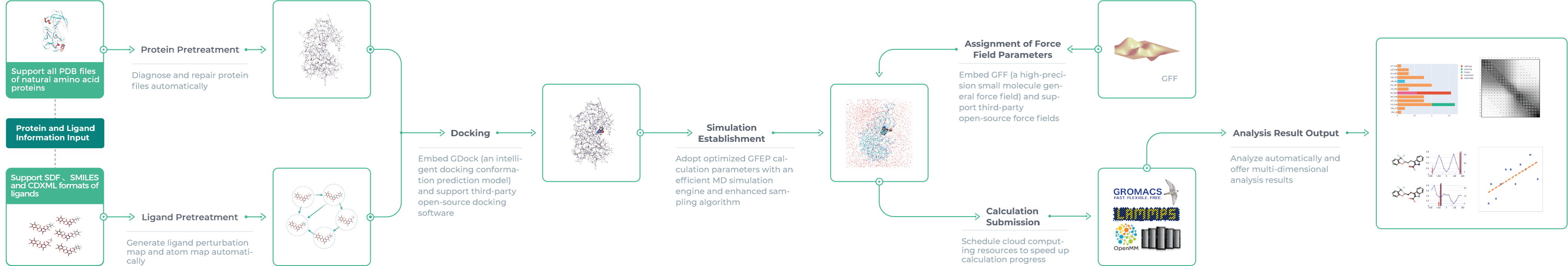
Types of Services


Predicting protein complex structures


Predicting target allosteric binding sites


Hit compound screening based on molecular docking


Compound activity screening and optimization based on free energy perturbation

Access to all cloud computing platform, enabling high-throughput calculation

We provide SaaS and private deployment based on customized needs

We accept different cooperation methods including annual subscription, fee by quota and co-development