



平台介绍
智能计算平台M1是星药科技自主研发的、面向药物发现和设计的AI驱动计算平台。其结合了人工智能与经典物理学原理,可快速准确描述分子和蛋白间的相互作用、精确计算目标药物分子与特定靶标的结合自由能,突破经典计算模拟方法,超越世界顶尖水平。
智能分子对接模块
基于星药原创的小分子靶蛋白结合构象及亲和力预测模型TBind开发的M1平台专有高通量对接模块
其以超越传统对接软件的效率和精度,从二维小分子结构准确预测得到三维对接结构,解决了基于靶点的药物设计中的关键问题,同时也可以为下游的自由能微扰计算提供合理的输入,从而进一步提升自由能微扰计算的预测精度。M1提供基于GDock的高通量分子虚拟筛选模块,能够在短时内完成百万/千万级分子的亲和力打分排序。MI也提供基于DynamicBind的动态对对接模块GDynamic,能够在对接过程中同时考虑蛋白柔性和动态,找到最适合输入分子的蛋白构象,让对接中发现隐藏口袋成为可能。
高精度分子力场模块
基于传统计算化学框架和星药原研人工智能算法开发的专门针对药物小分子的新一代通用力场
其以大量的量子化学第一性原理计算数据以及理化实验数据作为拟合目标,能够准确描述分子内、分子间相互作用,是高精度分子体系模拟计算的重要基础,也是基于结构的药物设计方法中的关键一环。
高性能自由能计算模块
基于人工智能和经典分子动力学开发的、结合星药自研力场GFF和对接模块GDock的自由能微扰计算模块
其解决了当前自由能微扰计算流程中的输入结构和力场精度两大痛点,可以更快速且更精确地计算目标药物分子与特定靶标的结合自由能,评估药物分子和蛋白靶点的结合强度,从而大幅提升药物研发关键环节的效率。

基于星药原创的小分子靶蛋白结合构象及亲和力预测模型TBind开发的M1平台专有高通量对接模块
GDock是全球首个能同时预测小分子和靶蛋白三维结合构象及结合亲和力的深度表征学习分子对接方法,其表现大幅超越现存方法的最好结果。
GDock采用端到端的数据驱动范式,结合物理启发式的几何图神经网络,打通了复合物三维结合模式及结合强度的双重预测,实现了对国际商业分子对接软件精度和效率的双超越,为分子蛋白相互作用预测提供了国产首个突破性方案。
GDock将Ligand RMSD小于5Å的比例从约30%提升至65%;对于结合中心的预测与真实中心距离小于5Å的比例更是从48%提升至82%。
由于模型摒弃了繁琐的传统采样方法,利用数据驱动的AI势能面进行结构生成,在预测和筛选的效率上也得到了大幅提升,全局对接的任务中每个分子仅需要0.5秒,是通用学术软件的四百分之一、知名商业对接软件的两千分之一。除了速度和精度的优势外,GDock还擅长捕捉非正构位点,能够处理传统Docking软件解决不了的非正构问题。

高通量对接
对接速度提升2000倍以上

识别非正构位点
GDock具备更强的全局预测能力,能够更好地捕捉非正构口袋的信息:
PRMT5蛋白拥有多个结合口袋,其新发布的PDB共晶结构6K1S发现了一个全新结合位点,GDock虽然从未见过结合该非正构口袋的小分子,但是仍然正确地定位到了真实结合的位置(而其他方法则更倾向于常见的正构位点) 。
Centroid Distance (Å) - global docking
GDock: 0.65
Academic Docking Software: 11.21
Commercial Docking Software: 27.36

基于DynamicBind的动态对接模块GDynamic,能够在对接过程中同时考虑蛋白动态的构象变化,找到最适合输入分子的蛋白构象,让对接中发现隐藏口袋成可能。

基于传统计算化学框架和星药原研人工智能算法开发的专门针对药物小分子的新一代通用力场

传统力场基于专家经验,对不同化学环境中的原子进行抽象和定义。为了进一步强化GFF对更大化学空间的描述能力,星药结合AI技术,使用图神经网络构建连续化的原子类型。全过程由平滑的神经网络函数构建,模型参数端到端可微分。基于神经网络构建的力场GFF可以轻松扩展和应用到任意分子,在包括能量预测和构象优化等一系列核心模块中突破传统力场的天花板。GFF拥有令人惊叹的精度、极强的泛化能力和优秀的迭代适配能力。
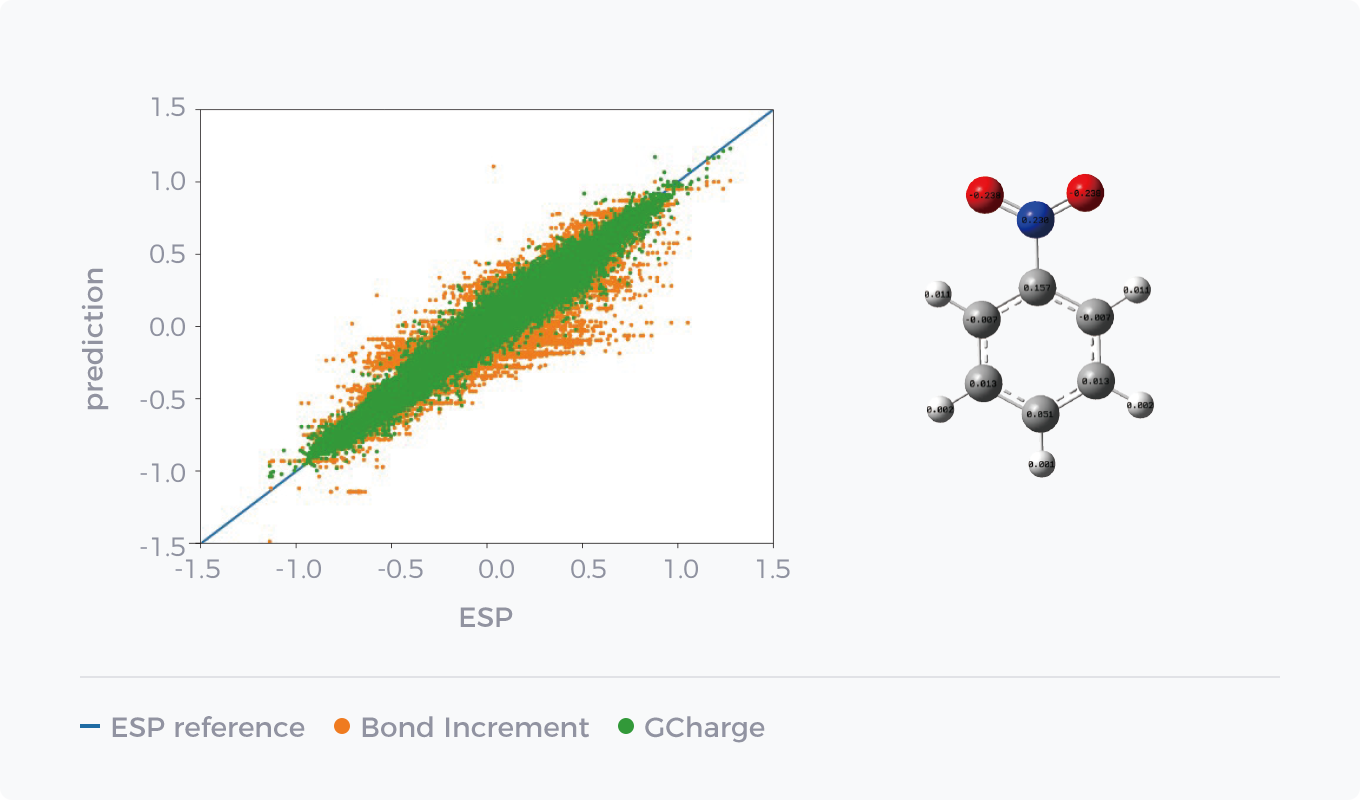
AI技术在非键参数的拟合方面起到极大的促进作用,其中一个典型的例证是我们基于图神经网络训练得到的GCharge电荷模型。其基于数百万QM计算数据作为训练集,相比于传统的非QM方法,可以更快速且准确地获取接近QM精度的电荷数据,显著提升了力场在水合自由能以及小分子结合自由能等性质上的预测精度。
- 能量预测
- 构象优化
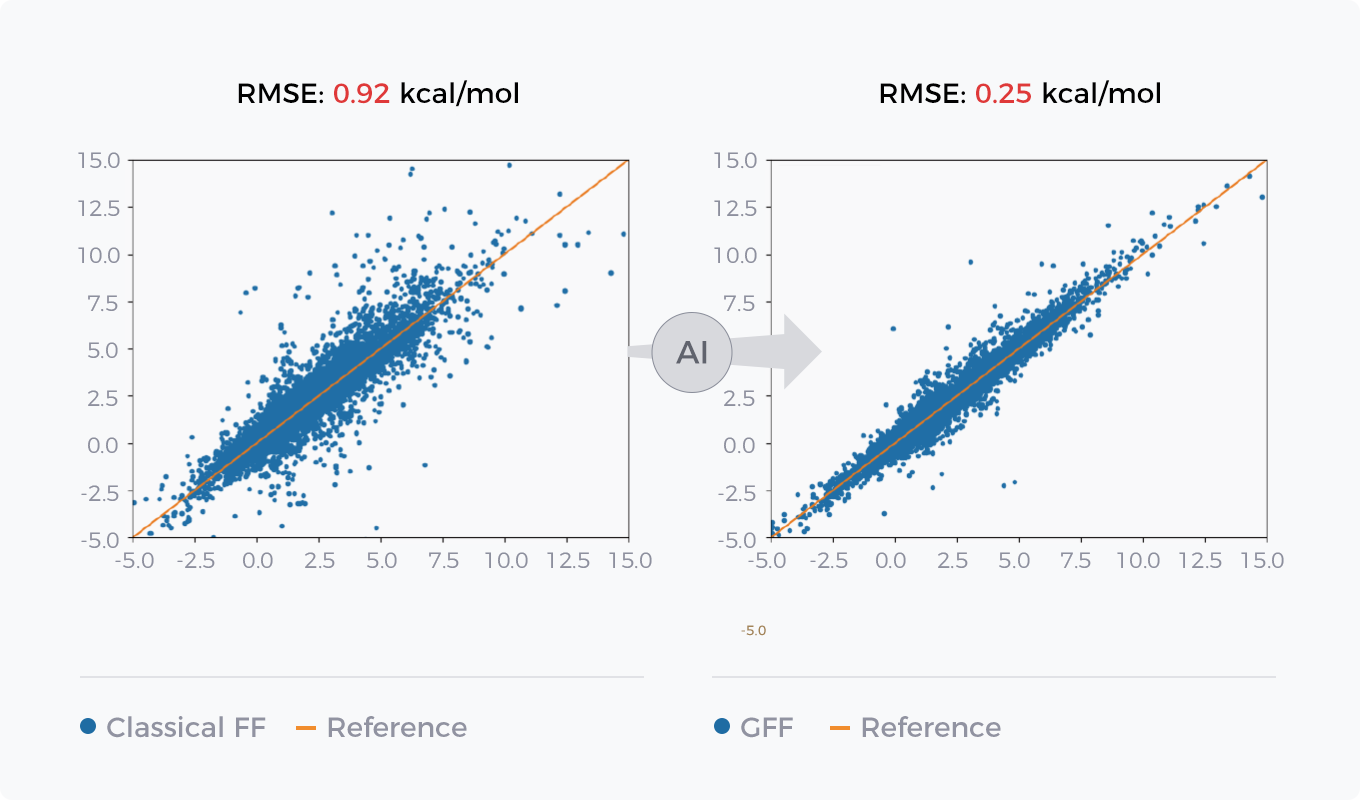
AI力场GFF与经典力场在能量预测及构象优化任务上的对比
AI技术在非键参数的拟合方面起到极大的促进作用,其中一个典型的例证是我们基于图神经网络训练得到的GCharge电荷模型。其基于数百万QM计算数据作为训练集,相比于传统的非QM方法,可以更快速且准确地获取接近QM精度的电荷数据,显著提升了力场在水合自由能以及小分子结合自由能等性质上的预测精度。
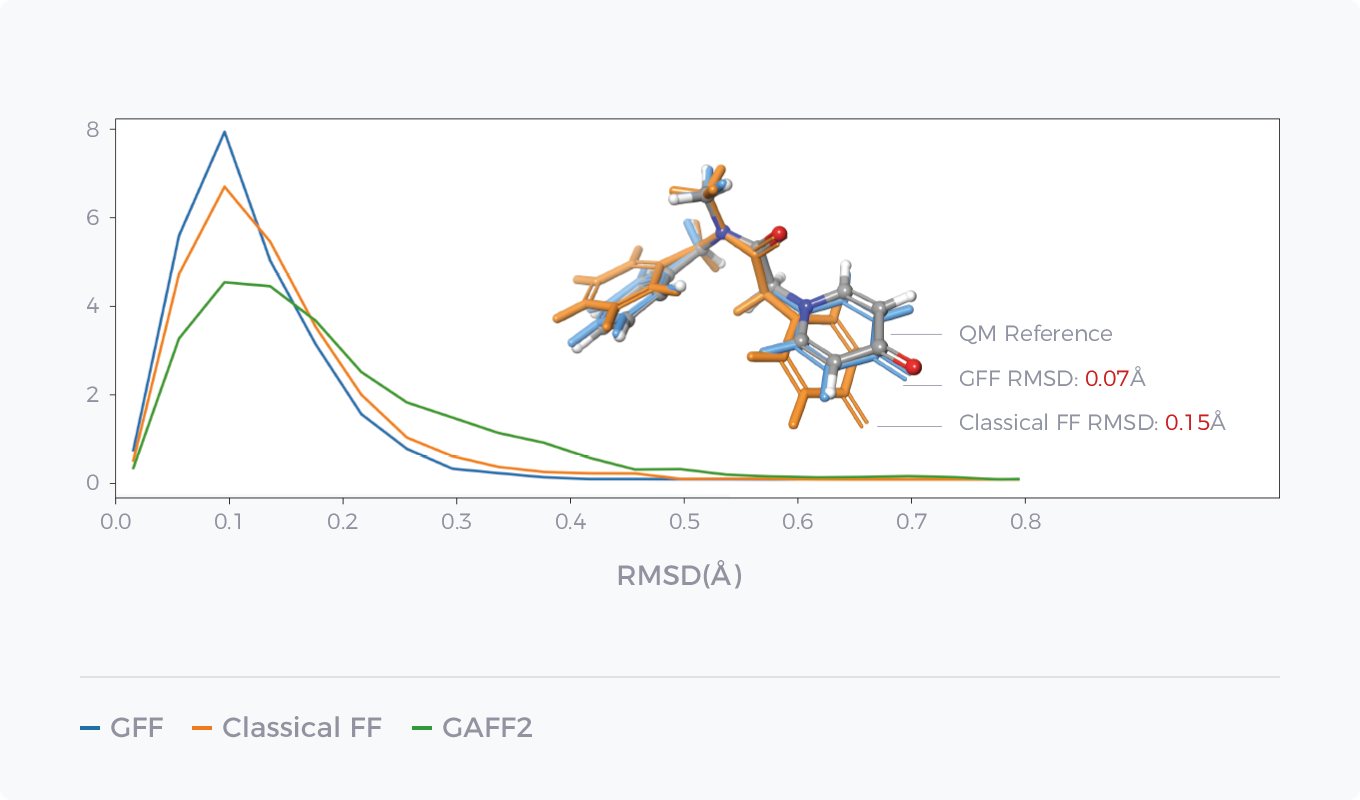
GCharge与传统电荷模型对比
- 水合自由能预测
- 结合自由能预测
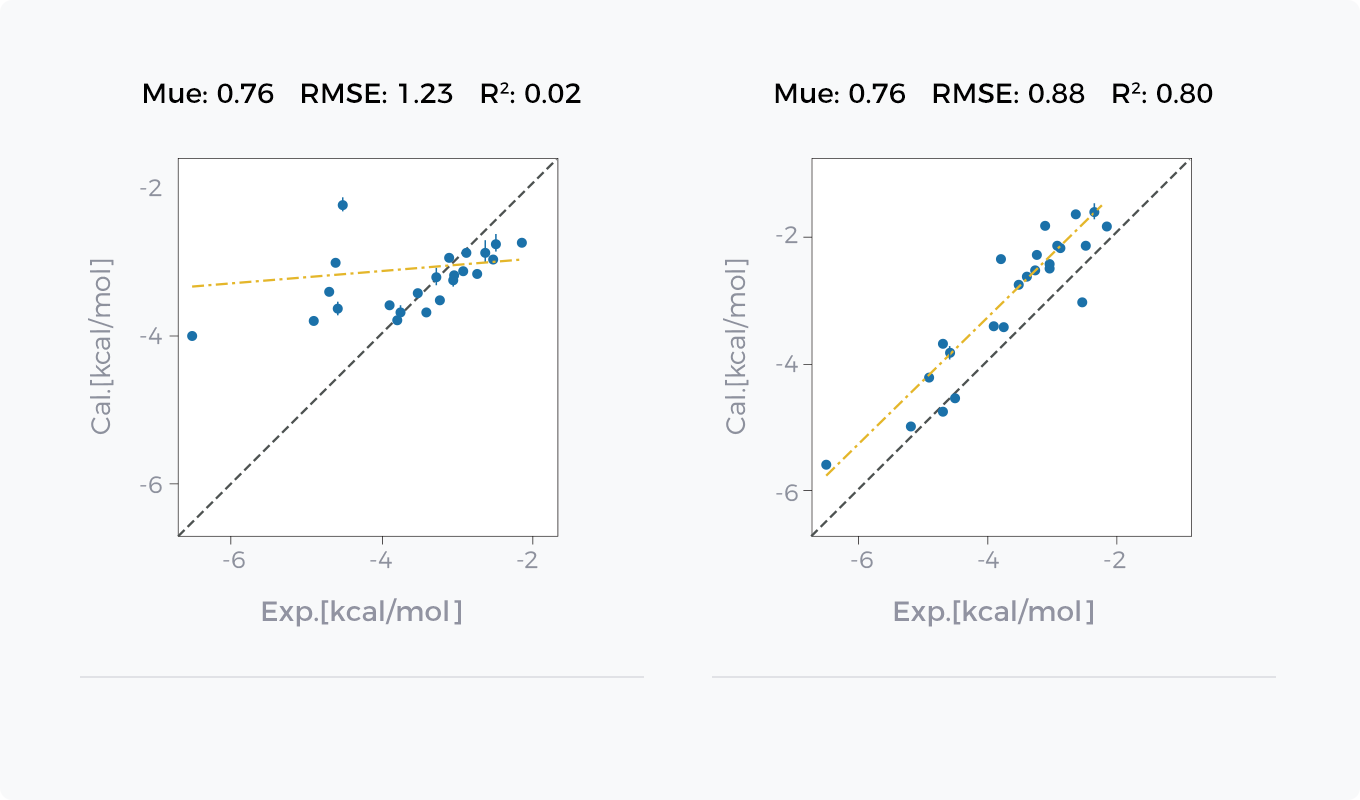

GCharge(右侧)与传统电荷模型(左侧)在水合自由能和结合自由能预测表现上的对比
GFF在能量预测、结构优化、势能面描述、理化性质预测、自由能计算等方面都表现出较高的精度,处于行业领先水平,显著优于绝大部分公开力场和商业力场。

基于人工智能和经典分子动力学开发的、结合星药自研力场GFF和对接模块GDock的自由能微扰计算模块
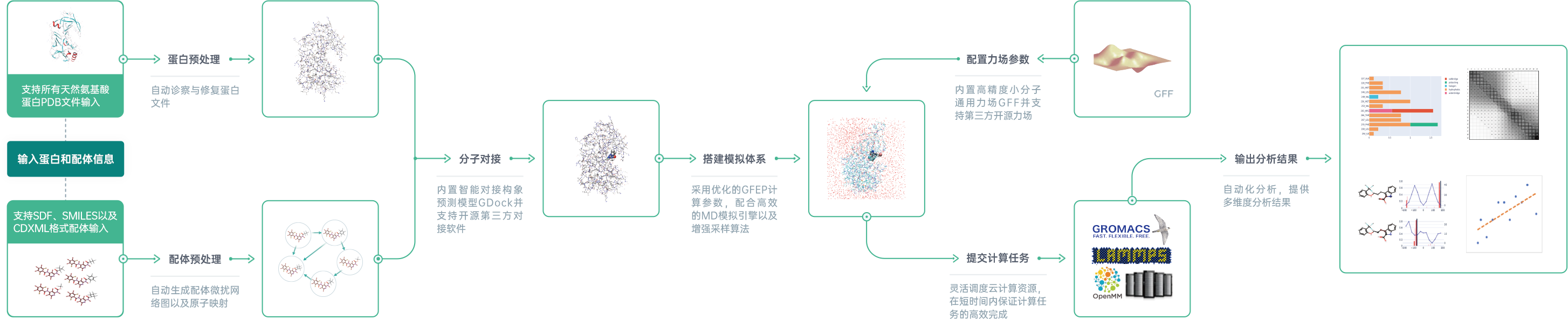
星药科技自主研发的基于人工智能的自由能微扰模块GFEP是针对药物研发领域自由能微扰需求的一站式计算平台。用户只需输入蛋白和配体信息,GFEP就能够自动完成结构优化、自由能微扰网络图构建、原子映射、分子对接、模拟体系搭建及力场参数配置等一系列分子动力学(MD)模拟的前处理步骤。所有模拟计算都会通过星药内部搭建的通道提交至超算云平台,通过调度可伸缩的计算资源执行计算任务,保证效率的同时也保证了数据的安全性。计算完成之后GFEP会产出包括结合自由能预测、相互作用分析、扭转角分析等结果在内的详尽分析报告。
业务类型


蛋白复合物结构预测


靶点异构结合位点预测


基于分子对接的苗头化合物筛选


基于自由能微扰的化合物活性筛选及优化

对接各大云计算资源
实现高通量计算

提供SaaS和私有化部署
满足不同需求

按年付费、按量付费、联合开发
等多种合作模式